AlienTrimmer v3 quickly and accurately filters out troublesome bases from nanopore sequencing read ends

Criscuolo (2026) EMBnet.journal 31, e1078 https://doi.org/10.14806/ej.31.0.1078
Received: 23 August 2025 Accepted: 14 January 2026 Published: 07 May 2026
Abstract
Since its first release in 2013, the sequence read trimming tool AlienTrimmer has been continuously improved, for use in various practical cases. In this context, the growing popularity of nanopore sequencing has made it necessary to update AlienTrimmer to deal with such long reads. New features were then implemented while guaranteeing that AlienTrimmer can still run fast. As illustrated in this note, the last release of AlienTrimmer can now filter out troublesome bases (i.e., low-quality and/or exogenous bases) from nanopore read ends with both first-rate accuracy and speed.
Availability: the source code of AlienTrimmer is available at https://gitlab.pasteur.fr/GIPhy/AlienTrimmer
Introduction
Nanopore sequencing is an increasingly used technology that enables the production of long reads at low equipment investment cost. Its widespread adoption in many omics areas goes hand in hand with notable improvements in the overall quality of the sequenced reads, e.g., increased lengths, lower error rates (Zhang et al., 2024). However, because of the ligation of technical oligonucleotide sequences (adapters, barcodes) during library preparations, such long reads can contain exogenous nucleotide segments at the ends. Moreover, base quality can often drop at both ends (Delaye and Nicolas, 2021). These troublesome ending regions should then be removed using trimming methods to avoid negative impacts in downstream analyses (e.g., de novo genome assembly, metagenomic characterisation).
Many tools exist to cut troublesome bases, but these approaches were initially implemented to specifically deal with short reads and generally require long running times (Chen et al., 2018). Novel programs were thus developed for nanopore sequencing reads, but they generally focus on low-quality bases only, such as Prowler (Lee et al., 2021). Furthermore, one of the few tools able to cut both low-quality and exogenous bases from such long reads (Porechop1) is unsupported since 2018. Hence, there arises a need for releasing complete trimming solutions able to deal with the widely adopted nanopore data.
AlienTrimmer was developed in 2013 at the Institut Pasteur (Paris, France) with the initial aim of implementing a method to detect and filter out any (alien) oligonucleotide (sub)sequences from read ends, with running times that are independent of the number of specified alien sequences. Based on the decomposition of each alien sequence into k-mers (to avoid any pairwise alignment), the Java program AlienTrimmer v1 was able to quickly process (in linear running time) large FASTQ files (Criscuolo and Brisse, 2013, 2014). AlienTrimmer v2 was released in 2020 with new features: the original k-mer matching method remained unchanged, but its updated purpose was to cut the longest read ends that contain a majority of bases of low quality and/or included into an alien k-mer. AlienTrimmer v2 still ran fast, owing to the continuous improvement of the Java Development Kit (JDK), as well as the emergence of efficient binary builders (e.g., GraalVM). In parallel, the “Alien” tool family has expanded with two accompanying programs: AlienRemover2 in 2021, to discard contaminating read and AlienDiscover3 in 2023, to infer alien sequences without any prior knowledge.
This technical note introduces AlienTrimmer v3 and its new features dedicated to nanopore data. AlienTrimmer v3 is shown to be able to perform accurate trimming of such long reads, provided that dedicated parameters are set. These performances are assessed by comparing it with fastplong, a long-read-dedicated version of the well-adopted tool fastp (Chen et al., 2018; Chen, 2023).
Materials, Methodologies and Techniques
Given a set of alien sequences, AlienTrimmer first decomposes each of them (and their reverse-complement) into alien k-mers of length k (option -k). Each input read is then traversed to detect every alien k-mer occurrence (Criscuolo and Brisse, 2013). During read traversal, all bases with a Phred score lower than a threshold Q (option -q) are also spotted. Troublesome bases are therefore those that are included into an alien k-mer and/or with a Phred score < Q. AlienTrimmer then searches for the longest prefix/suffix that consist of > 50% troublesome bases. During this searching step, AlienTrimmer tolerates up to a fixed maximum number of successive non-troublesome bases (option -m). Next, every delineated troublesome prefix/suffix is cut. Finally, each processed read is discarded when its length is lower than a given cutoff (option -l) or when it still contains a too important percentage of low-quality bases (option -p).
By default, AlienTrimmer is released with preset options for short reads, i.e. -k 10 -m 9 -q 13 -p 50 -l 50. But these parameters often result in inaccurate trimming when used on nanopore sequencing reads. However, different analyses (not shown) have led to the implementation of dedicated parameters, i.e. -k 9 -m 20 -q 13 -p 50 -l 500. Indeed, the use of slightly shorter k-mers (-k 9) and more tolerated successive non-troublesome bases (-m 20) generally yield accurate trimming in practice (see Results). Preset length cutoffs are arbitrary (-l 50 and -l 500 for short and long reads, respectively) and can be modified according to the user’s needs. Overall per-read quality can also be increased by lowering the percentage of accepted low-quality bases (option -p).
In addition, AlienTrimmer v3.2 complements the length- and quality-based filtering criteria (options -l and -p, respectively) with a third one (option -d), which is triggered when a read is likely to contain an internal alien (sub)sequence (i.e., outside its prefix/suffix), often indicative of a chimera. Their occurrences are assessed by read positions with a maximal alien k-mer coverage depth. Indeed, when position i reaches this maximal coverage depth, it is included in k successive alien k-mers, entailing the read substring [i−k+1, i+k−1] to match with an alien sequence (Criscuolo and Brisse, 2013). When option -d is set, AlienTrimmer v3.2 discards every read containing more than k positions with a maximal alien k-mer coverage depth.
Results
To illustrate its usefulness, AlienTrimmer was used to process a datafile (accession ERR8958607) derived from a nanopore sequencing of the Escherichia coli CFT073 genome (biosample SAMEA13167420; for more details, see Sanderson et al., 2024). After discarding reads shorter than 500 bases, the dataset consisted of 322,941 reads (2,236,910,669 bases). To assess the requirement for trimming these latter reads, they were aligned against the corresponding genome assembly (accession AE014075) using minimap2 v2.28 (Li, 2018) with option -x lr:hq, yielding ninit = 2,130,948,105 nucleotide matches and cinit = 37,054,290 clipped bases (CIGAR operators = and S, respectively). Secondary alignments were not considered, as well as chimeric ones (SAM tag SA). Yet the important number cinit of clipped bases confirmed the trimming requirement.
AlienDiscover v0.3 was first run to infer putative alien sequences (options -f 200 -x -c 50), leading to four prefixes and 11 suffixes (see Supplementary File4). Prefixes consisted in three low-residue sequences (i.e., heteropolymers poly-AT, -CG, -GT) and one technical sequence derived from an Oxford Nanopore Technologies Rapid Barcoding Kit (i.e., barcode RB02 with flanking sequences). Suffixes were only low-residue sequences (i.e., all four homopolymers, and the heteropolymers poly-AC, -AG, -AT, -CG, -CT, -GT, -CGG).
AlienTrimmer v3.2 was run with the 15 inferred alien sequences and option -N (equivalent to: -k 9 -m 20 -q 13 -p 50 -l 500), leading to 315,042 reads (2,185,276,189 bases). For matters of comparison, fastplong v0.4.1 was also used with the same 15 sequences (option -a) and low-quality base trimming options (i.e., -5 and -3), yielding 311,785 reads (2,124,199,782 bases). For a fair comparison, both tools were run again with comparable settings, i.e., length cutoff of 50 bases and no read discarding based on overall low quality. Under these settings, AlienTrimmer (-k 9 -m 20 -q 13 -p 100 -l 50) returned 322,941 reads (2,188,638,494 bases), whereas fastplong (-5 -3 -Q -l 50) returned 323,044 reads (2,200,690,042 bases). AlienTrimmer therefore removed more bases than fastplong (i.e., 2.16% and 1.62%, respectively). However, after aligning the trimmed reads against the genome assembly, AlienTrimmer yielded n = 2,120,255,217 nucleotide matches and c = 876,042 clipped bases, while fastplong led to n = 2,128,530,714 and c = 4,254,722. AlienTrimmer then detected far more troublesome bases than fastplong (4.85× smaller c), but at the cost of a slight loss of non-exogenous bases (1.004× smaller n).
For further comparison, AlienTrimmer (-p 100 -l 50) was run with varying parameters, i.e., k = 8, 9, …, 13, m = 10, 15, …, 35 and q = 7, 10, 13, 20 (options -k, -m, -q, respectively), as well as fastplong (-5 -3 -Q -l 50), i.e., d = 0.10, 0.15, …, 0.45 (option -d to set the upper threshold of the dissimilarity between an alien sequence aligned against a read region), M = 7, 10, 13, 20 (option -M to set the threshold Q of the average Phred quality within a sliding window) and W = 1, 4, 7, 10, 30, 50 (option -W to set the sliding window size; for more details, see documentation of fastplong5). Each returned read set was aligned against the genome assembly to obtain the numbers n and c of nucleotide matches and clipped bases, respectively (see Supplementary File6). The ability of performing an accurate trimming was assessed by estimating true positive outcomes as TP = cinit − c (i.e., number of initially clipped bases that have been removed), false negative as FN = c (i.e., number of remaining clipped bases), true negative as TN = n (i.e., number of matching nucleotides after trimming) and false positive as FP = ninit − n (i.e., number of initially matching nucleotides that have been removed). The overall accuracy of each tool was characterised by its ability to simultaneously maximise the true positive rate TPR = TP / (TP + FN) and minimise the false positive rate FPR = FP / (FP + TN). Corresponding receiver operating characteristic (ROC) plots are represented in Figure 1.
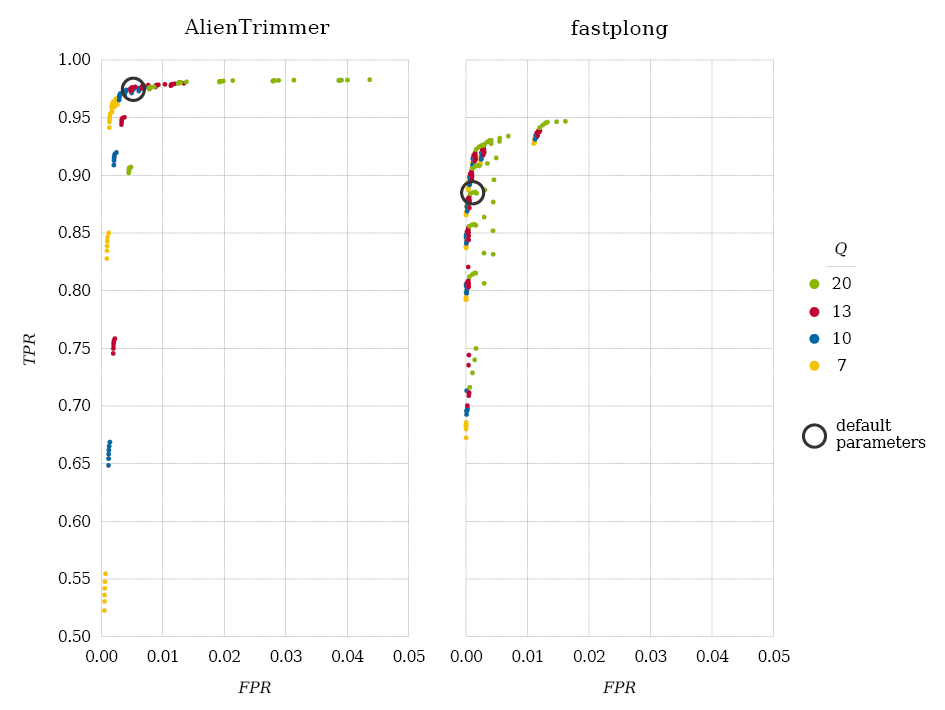
Figure 1. ROC plots showing the ability of AlienTrimmer v3.2 (left) and fastplong v0.4.1 (right) to perform accurate troublesome base trimming (i.e., high TPR and low FPR) when used with various parameters. The impact of Phred score thresholds Q is illustrated using coloured dots, i.e., Q = 7 (yellow), 10 (blue), 13 (red), 20 (green). Results obtained with the preset parameters of each tool are highlighted with a black circle. Note the different scales for FPR (x-axis from 0.00 to 0.05) and TPR (y-axis from 0.5 to 1.0).
The ROC plots show that the ability of the trimming tools to accurately remove troublesome bases is strongly associated with the quality cutoff Q under which low quality sequencing is assessed. Although AlienTrimmer and fastplong use different criteria to determine a low-quality region (i.e., global proportion and local average, respectively), they both failed at cutting enough bases (e.g., TPR < 97%) when Q was small (e.g., Q = 7). In Figure 1, bottom left dots for AlienTrimmer and fastplong were obtained with options -k 13 -m 10 -q 7 and -d 0.10 -W 50 -M 7, respectively, and top right dots with -k 8 -m 35 -q 20 and -d 0.45 -W 50 -M 20, respectively (see Supplementary File7). Then, as expected, both TPR and FPR increased when using relaxed parameters (i.e., small k, large m and q; large d, W and M). Interestingly, AlienTrimmer reached higher TPR than fastplong (e.g., > 95%), confirming its ability to detect more troublesome bases. Of note, both tools can yield non-negligible FPR (e.g., > 1%) when setting too relaxed parameters (e.g., Q = 20), showing that fine-tuned parameters are required to reach a good balance between high TPR and low FPR. AlienTrimmer recommended options (-k 9 -m 20 -q 13) for nanopore data yielded TPR = 97.6% and FPR = 0.5%, whereas default fastplong ones (-d 0.25 -W 4 -M 20) led to TPR = 88.5% and FPR = 0.1%. However, fastplong was able to perform a better trimming when using relaxed dissimilarity upper thresholds (e.g., TPR = 94.3% and FPR = 1.2% with -d 0.45 -W 4 -M 20).
Surprisingly, AlienTrimmer was observed to run faster than fastplong, despite using one and 16 threads, respectively. However, it is worth noting that the programming language of AlienTrimmer, Java, is also a key feature, as it can be compiled and executed in a variety of ways, yielding different execution speeds (Criscuolo and Brisse, 2014). This point was assessed by compiling the source code of AlienTrimmer v3.2 to create (i) a Java executable JAR file (using OpenJDK v25.0.18), and (ii) a native executable (using native-image v25.0.1 from the GraalVM JDK9). Using the same 15 alien sequences, the two AlienTrimmer executables were run (option -N) several times, as well as fastplong (-5 -3) on three (default) and 16 threads, to finally represent the averaged running times (Figure 2). Clearly, the AlienTrimmer executable built using native-image ran very quickly, up to 10× faster than the JAR one, and from 1.3× to 5× faster than fastplong (using 16 and three threads, respectively). Of note, writing compressed FASTQ files can have a negative impact on the overall running times of both tools, although fastplong seems less affected than AlienTrimmer (Figure 2).
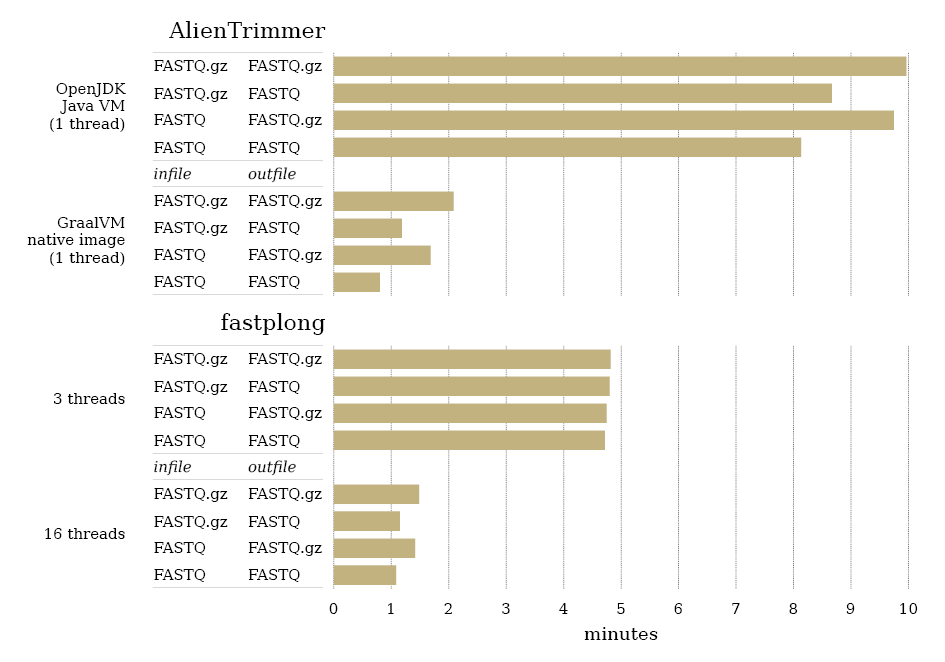
Figure 2. Running times of AlienTrimmer v3.2 (top) and fastplong v0.4.1 (bottom) measured in minutes on the same computational unit (AMD EPYC 7552 2.2GHz 48-core CPU; 500Gb RAM). Two different AlienTrimmer executables were used (each being single-threaded), whereas fastplong was run on three and 16 threads. For each computing case, running times were measured for each of the four combinations of compressed/uncompressed FASTQ-formatted infile/outfile.
Discussion
The last released version 3.2 of AlienTrimmer can filter out troublesome bases from nanopore sequencing reads with excellent accuracy and speed, while requiring low computational resources. When run on a real-case dataset, AlienTrimmer reached the 5% error zone (i.e., TPR > 95% and FPR < 5%), unlike the alternative program fastplong. Moreover, when purposely compiled, AlienTrimmer can also reach very fast running times, making it a useful tool to carry out a standard preprocessing step when dealing with sequencing data.
Acknowledgements
The author thanks Laetitia Fabre for her useful comments on the manuscript and Sylvain Brisse for continuous support. The author is also grateful to the anonymous reviewers for their comments. This work used the computational and storage services provided by the IT Department at Institut Pasteur.
Key Points
- • Removal of troublesome bases (i.e., low-quality and/or exogenous bases) from raw read ends often improves the quality of results from downstream analyses.
- • The new release of AlienTrimmer comes with preset parameters dedicated to nanopore data.
- • AlienTrimmer’s new features enable it to accurately filter out troublesome bases from both ends of nanopore sequencing reads.
- • AlienTrimmer still runs fast when purposely compiled.
References
- Chen S. Ultrafast one-pass FASTQ data preprocessing, quality control, and deduplication using fastp. iMeta. 2023;2:e107. https://doi.org/10.1002/imt2.107
- Chen S, Zhou Y, Chen Y, Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34(17):i884-90. https://doi.org/10.1093/bioinformatics/bty560
- Criscuolo A, Brisse S. ALIENTRIMMER: A tool to quickly and accurately trim off multiple short contaminant sequences from high-throughput sequencing reads. Genomics. 2013;102(5-6):500-506. https://doi.org/10.1016/j.ygeno.2013.07.011
- Criscuolo A, Brisse S. AlienTrimmer removes adapter oligonucleotides with high sensitivity in short-insert paired-end reads. Commentary on Turner (2014) Assessment of insert sizes and adapter content in FASTQ data from NexteraXT libraries. Front Genet. 2014;5:130. https://doi.org/10.3389/fgene.2014.00130
- Delahaye C, Nicolas J. Sequencing DNA with nanopores: Troubles and biases. PLoS ONE. 2021;16(10):e0257521. https://doi.org/10.1371/journal.pone.0257521
- Lee S, Nguyen LT, Hayes BJ, Ross EM. Prowler: a novel trimming algorithm for Oxford Nanopore sequence data. Bioinformatics. 2021;37(21):3936-7. https://doi.org/10.1093/bioinformatics/btab630
- Li H. New strategies to improve minimap2 alignment accuracy. Bioinformatics. 2021;37(23):4572-4. https://doi.org/10.1093/bioinformatics/btab705
- Sanderson ND, Hopkins KMV, Colpus M, Parker M, Lipworth S, Crook D, et al. Evaluation of the accuracy of bacterial genome reconstruction with Oxford Nanopore R10.4.1 long-read-only sequencing. Microb Genom. 2024;10(5):001246. https://doi.org/10.1099/mgen.0.001246
- Zhang T, Li H, Jiang M, Hou H, Gao Y, Li Y, et al. Nanopore sequencing: flourishing in its teenage years. J Genet Genomics. 2024;51(12):1361-74. https://doi.org/10.1016/j.jgg.2024.09.007