An ontology based query engine for querying biological sequences
Received 5 July 2013; Accepted 10 September 2013; Published 14 October 2013
Competing interests: the authors have declared that no competing interests exist.
Motivation and Objectives
This work presents the design and proof of principles application of Boinq, a flexible query engine for querying and analysing sequence data based on bio-ontology based annotations.
The bandwidth of sequence data generation has increased spectacularly since the advent of so-called next generation sequencing techniques, now approx. eight years ago (Metzker, 2009). This rate is still increasing today due to single molecule techniques, which are expected to increase data rates even further (Blow, 2008). These developments have spawned development of data processing workflows. At some stage, these pipelines result in a set of reads from the instrument, mapped to a reference genome assembly.
In many applications (such as RNASeq or ChIPSeq) counting these reads in a certain region is the start of further analysis. In research environments, these private read data are combined with publicly available datasets to perform numerous integrative queries over dispersed and highly heterogeneous datasets. An example of such questions is to compare read counts between treatment A and treatment B in regions upstream of genes annotated with a certain gene ontology (GO) term. Such an analysis requires counting reads from two data sources, consulting the GO, and finding gene annotations from a public database. Such analyses still requires hacking together a combination of queries, data conversions and ad hoc scripts. This is time consuming and error prone, and furthermore requires specialised personnel.
An analysis of a set of example questions revealed that there is a need for a rapid analysis pipeline to:
- quickly specify and visualise regions of interest based on a number of criteria. In these criteria, interoperability with bio-ontologies is required;
- perform simple aggregating or ranking analyses in these regions.
The boundary condition imposed by working with a collection of distributed, heterogeneous data is the natural ecosystem for semantic web technologies. This fact, and the required interoperability with bio-ontologies led to the decision to leverage semantic web technologies for disclosing, integrating and querying sequence data. The use of semantic web technologies offers clear advantages. As sketched above, the technologies are ideally suited to deal with distributed, heterogeneous data sources, and a growing body of (molecular) biological knowledge is being disclosed using well defined bio-ontologies.
Drawbacks can be identified as well. First, the inherent freedom associated with exposing data as RDF creates challenges. Obviously, using a common technology is not sufficient for guaranteeing interoperability. Therefore, a way to describe data sources that describe annotation features needs to be agreed upon. While such an initiative needs discussion in a wider group, some minimum requirements for such a standard are put forward in what follows. A second drawback originates from the complexity for the layman to create queries for a liberal data space. For this reason, we felt the need to include a query builder into the platform, as discussed further on.
Methods
The Boinq framework is composed of the following components:
- an RDF store with SPARQL endpoint documenting available data sources for features. An ontology is available for this meta dataset and is discussed further;
- a set of local SPARQL endpoints for exposing feature sets from various sources as RDF data, either directly or through mapping of the underlying SQL data. An endpoint is available for querying a subset of a locally running ensembl core data set for homo sapiens (Flicek et al., 2012);
- an interface for exploring the feature data sources. It contains a visualisation component for inspection of the data using a built-in genome browser, and an interface for defining the analysis that needs to be done. The analysis is split up in two steps:
- definition of a region of interest;
- definition of the analysis (still under construction).
The Boinq application is offered as a web application, and is built on a Java software stack. The following technologies were used to develop the framework:
- the client interface is built using SmartGWT[1];
- ontologies were built using Protégé[2];
- the server software is composed of individual components orchestrated using Spring[3], and persistence is achieved using Hibernate[4];
- the RDF data and the ontologies used are exposed as a SPARQL endpoint using the Apache Jena framework[5], more specifically the fuseki component. This framework is also used as a SPARQL client;
- mapping relational data to RDF dynamically is done using d2rq[6] (Bizer & Seaborne, 2004). The mapping from the ensembl core to FALDO[7] is documented on-line;
- Apache tomcat is used as application server;
- asynchronous jobs are handled using Quartz[8].
The architecture is depicted graphically in Figure 1.
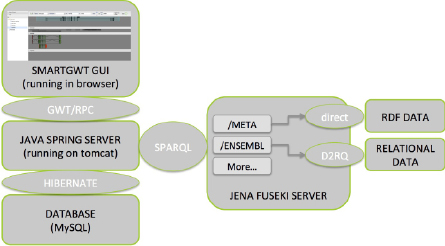
Figure 1. Architecture of the boinq application.
Results and Discussion
The boinq tool in its current version assumes the presence of data sources providing genome annotations (or features) through a SPARQL endpoint. We limit our application to features that are mapped to a publicly available reference genome and with exactly known positions on this reference.
Describing RDF feature data sources
Currently, exposing features as RDF data is not yet common practice, yet several initiatives are in place for doing just this in the near future. As common standards are not yet in place, we define in this section the assumptions about a feature data source. We have tried to make use of publicly available ontologies as much as possible:
- the position of a feature on a reference assembly is described using FALDO[9];
- the types of features are described using the sequence ontology (Eilbeck et al., 2005).
All further information about feature types that are offered by a data source are described in the meta dataset, that is an RDFS/OWL repository, available at the boinq website. A direct SPARQL endpoint to this meta dataset is also available. In the current version, we are using a central repository, but we solicit cooperative efforts to ensure that each data source adheres to a common ontology describing its fields and feature types.
In addition to providing sufficient information about the resource for human interpretation, the data source RDF store needs to provide assistance to the region builder component (see further). It needs to detail the available (filterable) fields for each feature type, and their target data types. The query builder uses SPARQL to query the data source metadata repository.
The region builder
While SPARQL is relatively straightforward to use for a technical audience, the tool is intended for biologists, not computer scientists. In addition, querying pure RDF without a reasoner in place will fail to return desired results. Therefore, a query builder was designed to aid the user in defining “Match trees”, a combination of match operators that result in queries on SPARQL data sources for computing regions of interest.
An additional benefit of defining queries on a meta-level rather than directly in SPARQL is that generators can be built on top of the match tree that target other protocols for fetching remote features. For example, the design of the match tree is kept compatible with the DAS specification (Jenkinson et al., 2008), so a generator for DAS query URLs may be plugged in at a later stage.
The region of interest that is used to perform the analysis, is defined by setting a number of criteria on known features. The region of interest for the analysis is then composed of a set of regions bound to the set of matching features.
The criteria are built by combining Match operators. The following Match operators are currently implemented:
- Match Location - these are criteria regarding the location of the feature;
- Match Type - to only return features of the specified type;
- Match All - this is an aggregate operator that requires all subcriteria to match;
- Match Any - this aggregate operator requires at least one subcriterion to match;
- Match Field - this operator restricts the features to those that fit within a restriction on a property of the feature. This operator is discussed in further detail.
The match field operator makes extensive use of the data source metadata. Indeed, as RDF data have inherently little restrictions on data types or the type of statements that can be made, there is great freedom in the definition of SPARQL queries. In order to guide the user in defining queries that are relevant to the data at hand, some restrictions are necessary. The field match operator first queries, for the type of feature at hand, the fields that are known. For this, a SPARQL query is used that takes advantage of rdfs:domain statements on properties of superclasses of the feature type. To avoid a too wide selection however, superclasses are restricted to avoid generic classes such as owl:Class. For the property field that is chosen from this selection by the user, the rdfs:range statements are inspected for the target type of the selected property.
If no information is found, the user can enter either an URI or a literal of a supported type (string and some numeric types). If range statements do provide information about the target entity, there are two possibilities:
- the entity is further described in the data source metadata - in this case, the interface will recurs into the target entity, creating a subMatch on a field of the target entity, resulting in a chain of Matches;
- the entity is unknown - in this case, again the user can choose between a URI or a literal;
- the entity is a literal - in this case, the user can enter a restriction on the literal value.
- For literal values, the following restrictions are supported:
- strings will be interpreted as a regex that must match, and optionally be case insensitive;
- for numeric values, comparison operators can be constructed (=,<,>,…).
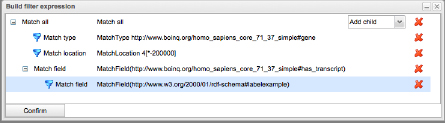
Figure 2. Example of a query under construction.
An example of a query under construction is given in Figure 2. In Figure 2, features of type “gene” are to be fetched form the ensembl Homo sapiens datasource, that have a transcript whos label matches “example”, and are located on chromosome 4, before position 200000. After construction of the Match tree, a SPARQL expression is generated that is used to fetch the set of matching features, and calculate the region of interest from that set. The user is presented the expression before use, so advanced users can make changes to the query, if necessary. For example, the query generated from Fig. 2 is
SELECT [...]
WHERE
{ ?feature
rdfs:label ?featureId ;
faldo:begin ?featureBegin .
?featureBegin
faldo:position ?featureBeginPos.
?feature
faldo:end ?featureEnd.
?featureEnd
faldo:position ?featureEndPos.
?featureBegin
faldo:reference ?featureRef.
?featureRef
rdfs:label ?featureRefName.
?featureBegin
rdf:type ?featurePositionType.
?feature
rdf:type ensembl:gene;
ensembl:has_transcript ?entity1.
?entity1
rdfs:label ?entity2 .
FILTER (str(?featureRefName) = “4”)
FILTER (?featureBeginPos <= 200000)
FILTER regex(str(?entity2),
“example”, “i”)
}
ORDER BY
ASC(?featureBeginPos)
External terms (terms from external ontologies) are handled in a specific way. The metadata repository specifies for each external term:
- a SPARQL endpoint;
- a SPARQL graph;
- optionally a term that is the superclass of any terms that can be used;
- optionally additional restrictions on the terms from the target ontology.
The system currently refers to Bioportal (Whetzel et al., 2011) for external terms. Upon encountering an external term, the user interface will use the Bioportal SPARQL endpoint to find and retrieve the relevant terms, and present them as a tree to the user. The user can also use full text search to find terms. The user has an option to retrieve all subclasses to be included in the match.
The visualization component
A built-in genome browser is available to inspect the computed regions visually. The browser is integrated in the interface and supports track based visualisation of generic features, and also shows ENSEMBL data as a background.
The analysis component
The analysis part is under development, but currently supports computing counting reads from a track in a predefined region of interest. Features to be included here are:
- ranking individual regions in a region of interest based on read count comparison. While further analysis is needed for validating findings based on these comparisons, it is already useful in focusing the effort;
- comparing read counts between tracks in a region of interest. For this, further research has to be performed into methods for computing statistical significance based on unknown data sources.
Availability
The tool is still under active development, but a prototype version is available at http://www.boinq.org/. This version currently supports basic queries based on Homo sapiens assembly v. 37. At this URL, you will find links to:
- a brief user manual
- the actual webapp
- the SPARQL endpoint
- the RDFS/OWL files
Acknowledgements
The work was made possible by a Baekeland scholarship from the Agency for Innovation by Science and Technology in Flanders (IWT). The first author furthermore wish to thank all Genohm colleagues, and in particular David De Beule and Ruben Simoens for their expertise and kind assistance with programming in SmartGWT and Spring.
References
Bizer C and Seaborne A. (2004) D2RQ-treating non-RDF databases as virtual RDF graphs. Proceedings of the 3rd International Semantic Web Conference, Hiroshima, Japan, November 7-11, 2004.
Blow N (2008) DNA sequencing: generation next-next. Nature Methods 5, 267-274. doi:10.1038/nmeth0308-267
Eilbeck K, Lewis SE, et al. (2005) The Sequence Ontology: a tool for the unification of genome annotations. Genome Biology 6(5). doi:10.1186/gb-2005-6-5-r44
Flicek P, Ahmed I, et al. (2012) Ensembl 2013. Nucleic Acids Research 41(D1), D48–D55. doi:10.1093/nar/gks1236
Jenkinson AM, Albrecht M, et al. (2008) Integrating biological data - the Distributed Annotation System. BMC Bioinformatics 9(Suppl 8), S3. doi:10.1186/1471-2105-9-S8-S3
Metzker ML (2009) Sequencing technologies - the next generation. Nature Reviews Genetics 11(1), 31–46. doi:10.1038/nrg2626
Whetzel PL, Noy NF, et al. (2011) BioPortal: enhanced functionality via new Web services from the National Center for Biomedical Ontology to access and use ontologies in software applications. Nucleic Acids Res. 39(Web Server issue), W541-545. doi:10.1093/nar/gkr469.